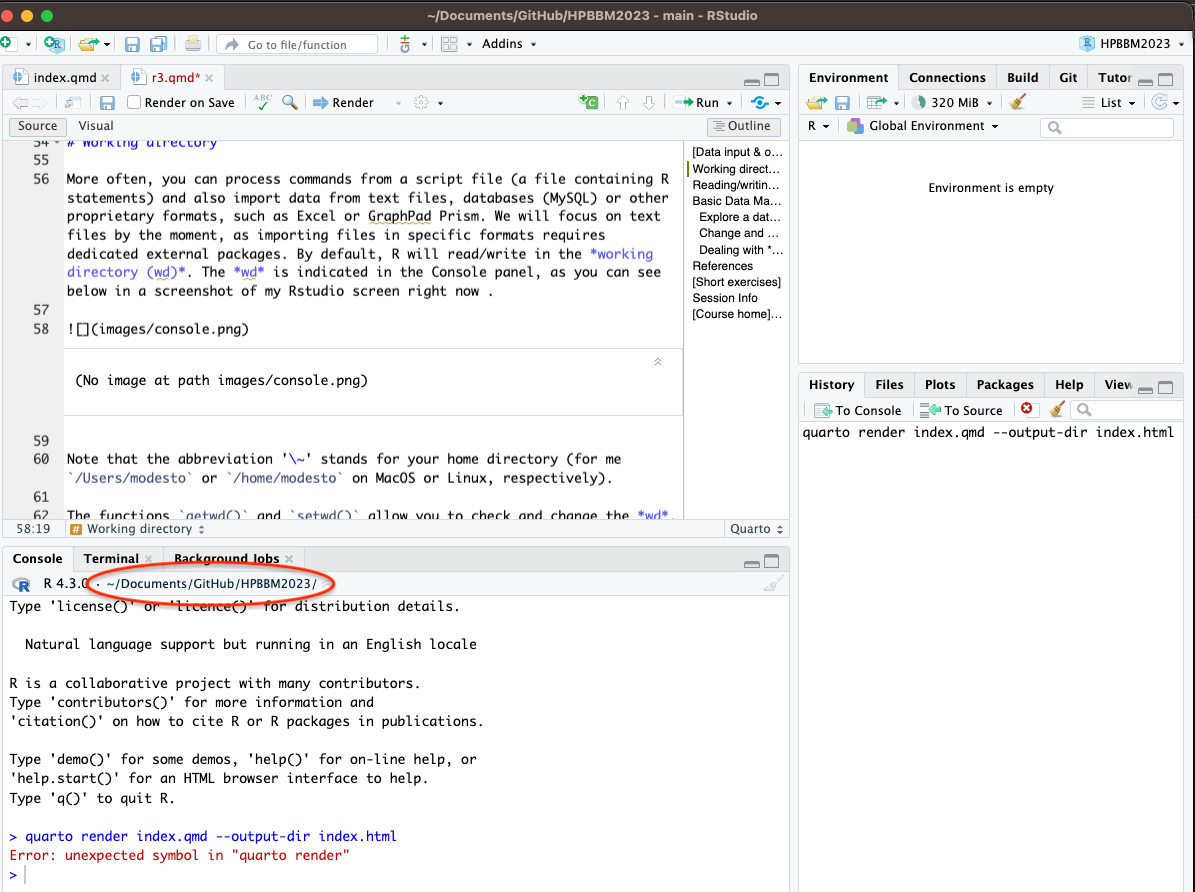
In most cases, you will use R to process also import data from text files, databases (MySQL) or other proprietary formats, such as Excel or GraphPad Prism and you will execute commands from a script file (a file containing R statements). We will focus on text files by the moment, as importing files in specific formats requires dedicated external packages. By default, R will read/write in the working directory (wd). The wd is indicated in the Console panel, as you can see below in a screenshot of my Rstudio screen right now.
Note that the abbreviation ‘~’ stands for your home directory (for me /Users/modesto or /home/modesto on MacOS or Linux, respectively).
The functions getwd() and setwd() allow you to check and change the wd. Try the examples below and pay attention to the output. Remember that you can write ?getwd() or ?setwd() for help.
In RStudio, the default working directory can be set from the “tools” and “global options” menu. Also, you can change the wd for your session in the menu Session > Set Working Directory and change it to that of source file (for instant your R script), or the selected directory in the files panel.
Wouldn’t it be great if you didn’t have to manually set the working directory every time?
If setting the working directory feels annoying or confusing, you can avoid it by using an R Project in RStudio, as explained in Lesson R7. R Projects automatically manage the working directory for you, making your workflow smoother and more reproducible.
1.1Quick exercise (I)
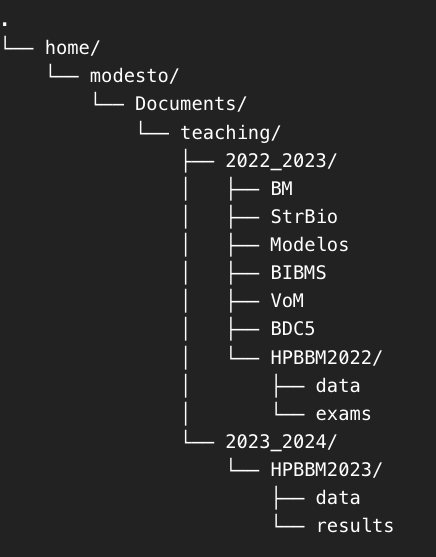
Let’s see if you understood. Consider the following tree of directories.
How would you change de wd to the folder HPBBM2023/data? You also can try with your own computer and your directory tree.
Select the right answer
2Reading/writing data in R
The most common way to read your data in R is importing it as a table, using the function read.table(). Note that the resultant object will become a Dataframe, even when all the entries got to be numeric. A followup call towards as.matrix() will turn it into in a matrix.
In the following example lines we read a file called small_matrix.csv, located in the data folder. If we attempt to make some matrix calculations, R will force the dataframe to a matrix when possible, but it will return an Error for many matrix-specific operations or functions unless, we transform the dataframe into a matrix.
sm <-read.table("small_matrix.csv", sep =",") #what happens here?
Warning in file(file, "rt"): cannot open file 'small_matrix.csv': No such file
or directory
Error in file(file, "rt"): cannot open the connection
Error in diag(sm): 'list' object cannot be coerced to type 'double'
# some operations cannot coerce a matrix but we can# explicit itdiag(as.matrix(sm))
[1] 2 10 13
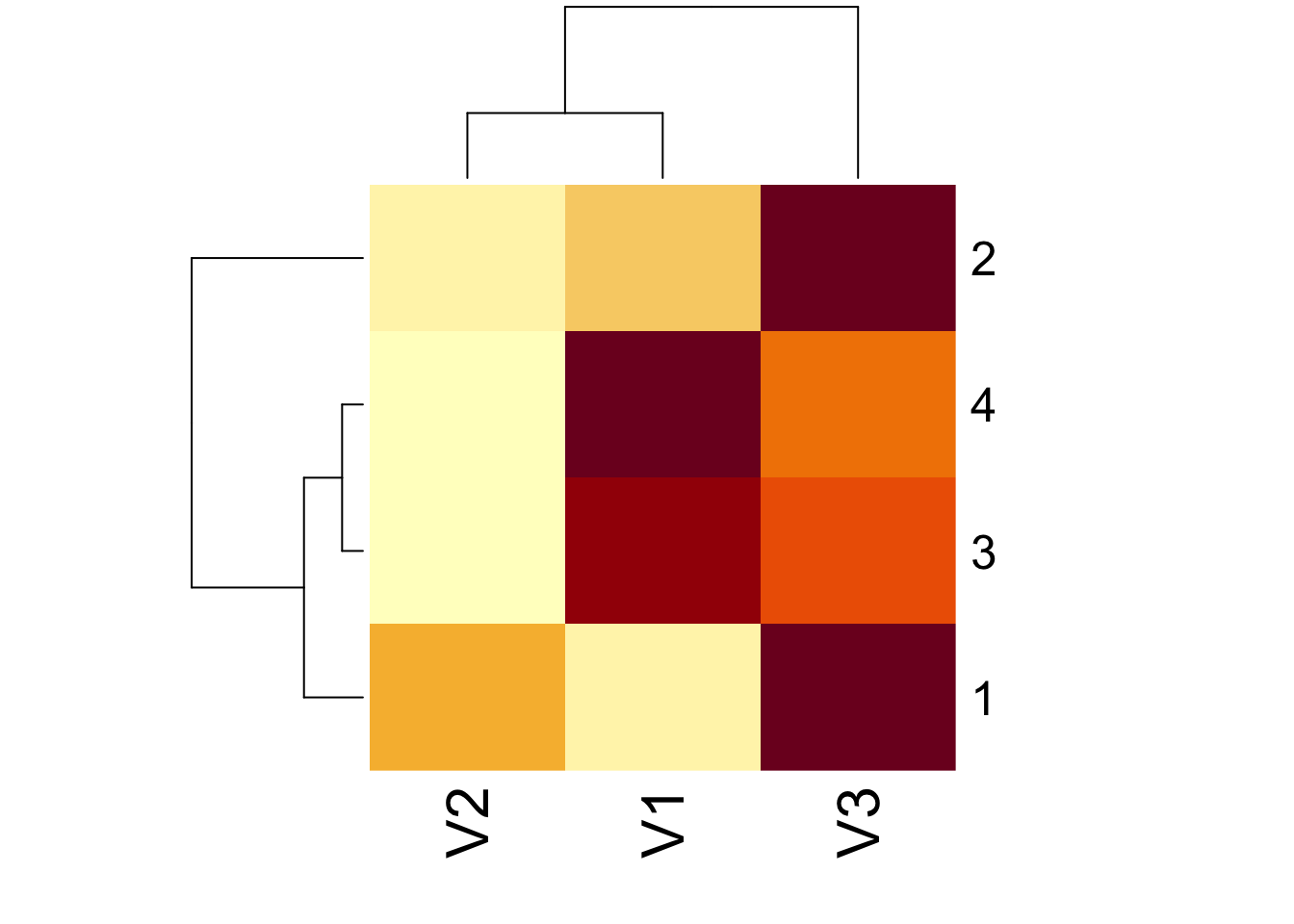
heatmap(sm)
Error in heatmap(sm): 'x' must be a numeric matrix
heatmap(as.matrix(sm))
Tip
Note that read.table() and its wrappers read.csv() or read.csv2() are mostly use for reading dataframes. If your want to read text files, like in the case of biological sequences, you can use other functions, like readLines(), described in the Lesson R8.
You can write any data object(s) as binary data file or as text files. Compare the different files saved in the code below.
Data files in RData format can be open from the Environment tab or with the load() function. To check that it was correctly loaded, we can remove it before loading with the rm() function.
# remove from the Global Environment in our RStudio sessionrm(vector)rm(vector2)# load againload("data/vector2.Rdata")vector
[1] 1 2 3 4 5
vector2
[1] 3 5 6 7 8 9 10 11 12 13
Caution
Note that if you save your data with save(), it cannot be restored under a different name. The original object names are automatically used. If you want to save the object and restore it with a different name you need to use the functions saveRDS() and readRDS()
2.1Quick exercise (II)
We are using the same directory tree. Your wd is HPBBM2022/data and you need to save a data.frame named table1 as table1.csv in the folder HPBBM2023/data using ; as separator. How would you do it without changing the working directory? Try it also with your own computer.
Select the right answer
[Interactive data input & output in R]
You will almost always import your working data from a file. However, especially if you are working with very small datasets, it is also possible to add your data interactively directly in the console, as you did in some of the examples before. You can also define your objects and enter your data interactively using the scan() and readline() functions, as in the examples below. For output, you can simply call the object by its name or use the print() function, which displays the contents of the object on the screen. You can try the following code.1
Although seldom used, you can also edit the contents of your objects using the function edit(). This function can be used to edit different objects, including vectors, strings, matrices or dataframes. MacOS users may need to install XQuartz X11 tool.
3Basic Data Management in R
Now we are going to import and explore an example dataset, containing metadata from an Illumina sequencing project of pathogenic E. coli strains (Flament-Simon et al. 2020, https://doi.org/10.1038/s41598-020-69356-6). However, for didactic purposes, the original data have been simplified and manipulated and the attached datasets do not fully correspond to the actual data. You can find the table in the data folder or download it in the following link: coli_genomes.csv.
3.1 Open and explore a dataframe
As you can see in the R help, the function read.table() has several default options as FALSE, like header=FALSE. When you have a spreadsheet export file, i.e. having a table where the fields are divided by commas in place of spaces, you can use read.csv() in place of read.table(). For Spaniards, there is also read.csv2(), which uses a comma for the decimal point and a semicolon for the separator. The latter functions are wrappers of read.table() with custom default options. Likewise, there are write.csv() and write.csv2(), which are wrappers of write.table(). Take a close look at the following examples that illustrate several ways to open a table — including some common mistakes — and how to explore it quickly.
# Note differences between read.table(), read.csv() and# read.csv2()coli_genomes <-read.table(file ="data/coli_genomes.csv")
Error in scan(file = file, what = what, sep = sep, quote = quote, dec = dec, : line 2 did not have 11 elements
Biosample Year.of.isolation Source
1 SAMN14278613 NA Human
2 SAMN14278614 2010 Human
3 SAMN14278615 2008 Human
4 SAMN14278616 NA Human
5 SAMN14278617 2011 Porcine
6 SAMN14278618 2007 Porcine
# explore the dataframe structuredim(coli_genomes)
[1] 25 16
length(coli_genomes)
[1] 16
ncol(coli_genomes)
[1] 16
nrow(coli_genomes)
[1] 25
# dataframe estructure in one linestr(coli_genomes)
Some of the columns include ‘chr’ data that may be actually a categorical variable, so we can code them as factor. Using the expression as.factor() you can check whether the data would correspond to a text or a categorical variable.
How many levels are there in Source?? It is not uncommon to see some mistake in our data, usually made when the data were recorded, for example a space may have been inserted before a data value. By default this white space will be kept in the R environment, such that ‘Human’ will be recognized as a different value than ‘Human’. In order to avoid this type of error, we can use the strip.white argument.
unique(coli_genomes$Source)
[1] Human Human Porcine Avian
Levels: Avian Human Human Porcine
[1] Human Porcine Avian
Levels: Avian Human Porcine
At this point, you may consider that writing the name of the dataframe every time that you want to work with it can be repetitive. In fact, we don’t need to do it.
attach(coli_genomes) #attachtable(Source)
Source
Avian Human Porcine
3 17 5
detach(coli_genomes)
Note that attach can be used for any R object, including dataframes, lists, vectors, packages… Once attached, R will consider those objects as databases, located in new, temporal environments.
Throw out the trash
If you want to remove some data to free up your computer’s memory, you can use the rm() function (aka remove()) to remove a specific object from your working environment. If you are running a script with large datasets, you can also use gc() to free memory after a large object has been removed. This is also done automatically without the user having to intervene, but it can be useful to call gc after a large object has been removed, as this can prompt R to return memory to the operating system.
3.2 Renaming, changing and adding variables
We can also rename some variables to use more easy names.
R has some built-in datasets that can be used as examples for plots or analysis. You can check all of them using the function data(). For the following exercise we are going to use one of that datasets, named quakes. You can import it as follows:
data(quakes)
Then, use the function str() to examine the structure of the quakes dataset. Which of the following best describes the variables represented in this data frame?
Select the right answer
Now you need to create a new dataframe quakes2 with the three columns, lat, long and new_var, being the latter the product lat\(x\)long. What is the mean of the new variable? Fill the gaps to finish your code in the R snippet below.
Load the file colis3.csv as colis and explore the dataset structure.
Calculate the mean of numerical variables: isolation date (Year), antimicrobial resistance genes (AMR), virulence factors (VF), CRISPR cassesttes (CRISPR), integron cassettes (Integron) and sequencing date (seqs) in those strains? Note. For the seqs variable you will need to use the function as.Date().
Save the tables coli_genomes_renamed and colis in a single Rdata file.
Add the values of the exercise 2 as a last row in the table. Nowyou will need to use the function format.Date() (or just format()) for the seqs variable.
Since it is interactive we cannot run it when we render the source file into a static web page. This can be prevented in markdown/quarto with the option `eval: false` in the chunk.↩︎